gpu服务器使用以及机器学习代码设置
希望本文能够帮助你高效利用课题组服务器的gpu资源,更好地完成学习科研任务
服务器连接部分
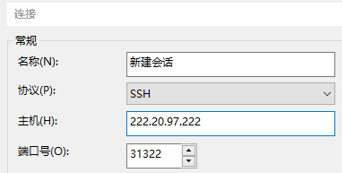
由于协议限制,slurm集群服务器无法使用Windows自带的远程桌面连接,需要下载支持SSH连接的软件,如“XShell”,“MoBaXterm”,输入指定的主机和端口号进行连接。(这里以xshell为例)
Slurm集群的主机号和端口号是222.20.97.222:31322
连接之后需要使用账号密码进行登录。如果没有账号密码可以找管理员寻求帮助。输入正确的账号密码之后即可成功连接
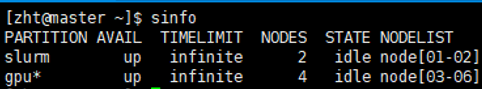
登陆成功之后显示如图所示,使用sinfo可以查看到当前有四个可用的gpu节点,分别是node03-node06
当前的gpu服务器的配置是四个独立节点,每个节点拥有8*4090显卡,即四个节点总共有32张4090显卡
环境配置部分
个人用户登录进来一般是处于master节点下的个人目录位置,可以在此创建子文件夹进行开发。
进行机器学习的有关前置步骤:十分推荐创建一个自己的虚拟环境,避免受到服务器本身配置环境的影响导致代码无法运行,或者对服务器环境造成影响。
简单理解什么是虚拟环境:
虚拟环境(Virtual Environment)是 Python 编程中的一个概念,它允许你为不同的项目创建独立的环境,每个环境可以有自己的依赖和库版本,而不会影响到其他项目。这就像是为每个项目准备了一个独立的“工具箱”,里面装有项目所需的特定工具和材料,而不会影响到其他项目的工具箱。
创建虚拟环境
构建虚拟环境的参考步骤:
虚拟环境创建:
使用python自带的venv模块创建虚拟环境,例如我现在想要在我的个人文件夹下面创建一个名叫python39venv的虚拟环境文件夹(也可以在其他文件夹下创建,更换路径即可):
python3 -m venv /public/home/zht/python39venv
激活虚拟环境:
source /public/home/zht/python39venv/bin/activate
在前面看见一个括号,括号中的名称为你的虚拟环境文件夹,即表明虚拟环境激活成功

退出虚拟环境:
Deactivate 即可直接退出虚拟环境
 从本地电脑上获取代码所需的依赖库
从本地电脑上获取代码所需的依赖库
这里假设你已经有了一个本地配置好环境的,可以运行的机器学习代码。在确保你已经激活你需要的响应环境之后,打开ide自带的控制台(这里以pycharm为例),使用命令pip freeze > requirements.txt即可生成一个有关的环境文件,例如我这里在路径下生成了一个名叫requirements1.txt的文件,后续可以用于配置环境
 以下是生成的requirements1.txt的文件的部分内容:
以下是生成的requirements1.txt的文件的部分内容:
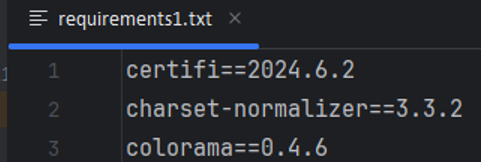 注意:请先确认你需要的python版本,虚拟环境是根据此python版本为基础进行设置的,一旦虚拟环境设置成功之后,不支持修改依赖的python版本,如果想修改python版本只能重新创建一个新的虚拟环境。
注意:请先确认你需要的python版本,虚拟环境是根据此python版本为基础进行设置的,一旦虚拟环境设置成功之后,不支持修改依赖的python版本,如果想修改python版本只能重新创建一个新的虚拟环境。
服务器上自带的默认python版本是3.9.18
依赖库安装
在创建好虚拟环境之后,即可进行相关依赖库的安装:
使用xftp等文件传输软件,把你在上一步生成的requirements.txt转移到服务器上你的文件夹中
首先激活虚拟环境,然后进入到requirements.txt所在的文件夹,使用pip install -r requirements.txt,即可安装相应的依赖包。如果你的依赖包比较多,可能需要较长的时间
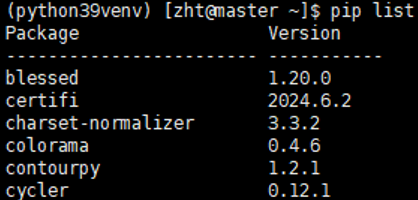
安装完成之后即可进行代码的运行。在激活虚拟环境之后,使用pip list即可查看当前安装的所有包
例如我查看当前我的虚拟环境下安装的依赖库所展示出的部分内容:
代码运行和日志文件输出
代码运行
那么在配置好运行环境之后,怎么运行代码呢?首先需要把代码放在服务器上面,然后需要一个运行脚本进行运行
例如我在服务器上,我的用户文件夹下面创建了mycode文件夹用于存放代码,里面有一个文件名为CNN.py,是我要运行的神经网络。然后我需要一个.sh脚本进行运行。下方即为脚本的主要内容。将脚本保存并且存放到服务器中,使用sbatch 即可进行执行!例如我这里的脚本名称叫cnn03.sh,切换到脚本目录下,使用命令sbatch cnn03.sh即可执行
#!/bin/bash
#SBATCH --job-name=run_cnn_node03 # 作业名称
#SBATCH --output=cnn_output_%j.txt # 标准输出文件
#SBATCH --error=cnn_error_%j.txt # 标准错误文件
#SBATCH --time=12:00:00 # 运行时间限制
#SBATCH --partition=gpu # 请求GPU分区
#SBATCH --nodes=1 # 请求1个节点
#SBATCH --ntasks-per-node=1 # 每个节点的任务数
#SBATCH --gres=gpu:8 # 请求1个GPU资源
#SBATCH --nodelist=node03 # 指定使用node03节点
# 激活虚拟环境
source /public/home/zht/python39venv/bin/activate
# 切换到存放Python脚本的目录
cd /public/home/zht/mycode
# 执行Python脚本
python CNN03.py
运行脚本:

服务器基本命令以及作业脚本等更多的内容可以查看本课题组网站内其他文章!
服务器日志输出
输出的日志文件默认是在和脚本文件同一目录下,在脚本文件里面可以设置输出文件和错误文件的名称。服务器会对提交的作业进行自动编号。如果使用类似这样的命名,那么输出文件这里的j就代表服务器自动编号的内容
#!/bin/bash
#SBATCH --job-name=run_cnn_node03 # 作业名称
#SBATCH --output=cnn_output_%j.txt # 标准输出文件
#SBATCH --error=cnn_error_%j.txt # 标准错误文件例如我提交之后显示的jobid为935
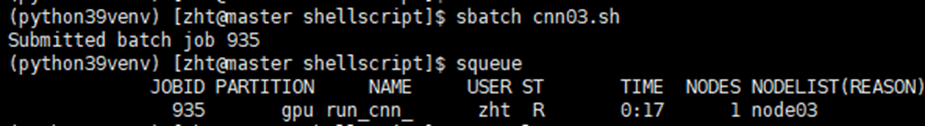 那么此时我这个作业的输出文件和错误文件的名称就是cnn_output_935.txt和cnn_error_935.txt
那么此时我这个作业的输出文件和错误文件的名称就是cnn_output_935.txt和cnn_error_935.txt
那么输出文件和错误文件有什么用呢?
错误文件会打印当前任务执行报错的内容,显示出程序无法正常运行的错误信息,便于进行python代码或者脚本程序的修改;而输出文件则打印出了当前运行的python文件中,你使用print输出的相关内容,便于查看运行过程中的一些内容
由于一个jobid对应的一个任务只会输出一个日志文件,那么如果有很多个并行,所有并行任务的输出内容都会输出到一个txt文件里面,并且不按照顺序进行写入,会导致日志的可读性极差。那么在这种情况下应该怎么办呢?一个暂时的解决方案是在输出的时候假如作业的pid,这样由于每个任务的pid都是独一无二的,就可以在日志里面进行区分了
import os
pid = os.getpid()
print(f"pid:{pid}, Total accuracy: {test_accuracies[-1]}")sbatch脚本配置相关内容
这里主要解释一些和机器学习有关的配置(其他配置看图片里的注释也可以大致知道什么意思)
既然我们有了高性能的gpu服务器,当然是希望能尽可能地把计算资源都利用上啦!但是如果不在作业脚本以及python代码中进行设置,可能并不能达到理想的效果
以下方一个我配置好的脚本为例,说明如何通过脚本调度服务器资源
#!/bin/bash
#SBATCH --job-name=cnn_node04 # 作业名称
#SBATCH --output=cnn_output_%j.txt # 标准输出文件
#SBATCH --error=cnn_error_%j.txt # 标准错误文件
#SBATCH --time=120:00:00 # 运行时间限制
#SBATCH --partition=gpu # 请求GPU分区
#SBATCH --nodes=1 # 请求1个节点
#SBATCH --nodelist=node04 # 指定使用node04节点
#SBATCH --gres=gpu:8 # 最多请求8个GPU资源
#SBATCH --ntasks=18 # 最多每个gpu上运行18个任务
#SBATCH --cpus-per-task=4 # 每个任务最多分配4个CPU核心
# 激活虚拟环境
source /public/home/zht/python39venv/bin/activate
# 切换到存放Python脚本的目录
cd /public/home/zht/mycode
n=20
for ((i=1; i<=n; i++))
do
srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN051.py &
srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN052.py &
srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN053.py &
srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN054.py &
srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN055.py &
srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN056.py &
srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN057.py &
srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN058.py &
wait
done
一些前置说明:
最上方大量的sbatch部分可以暂且理解为对脚本资源的请求与限制。
如果上方的请求内容和下方srun启动命令所申请的资源的数量不对等,那么服务器会优先使用下方的资源进行分配
Gpu分区每个节点的配置:
每个节点有两个cpu,每个cpu有36个核心,即共计72个cpu核心
每个节点的内存大约是1T
每个节点有八张4090
代码说明部分
#SBATCH --nodelist=node04 # 指定使用node04节点
#SBATCH --gres=gpu:8 # 最多请求8个GPU资源
#SBATCH --ntasks=18 # 最多每个gpu上运行18个任务
#SBATCH --cpus-per-task=4 # 每个任务最多分配4个CPU核心#SBATCH --nodelist=node04表示请求的节点,gpu部分总共有node03,node04,node05,node06四个节点,但是node03存在大量未知任务占用显存,所以如果利用全部资源跑大量并行任务,尽量在后三个节点运行。
#SBATCH --gres=gpu:8表示限制你的作业最多使用8个gpu资源,因为节点只有八个gpu,如果这个地方设置多了会无法运行并且报错
#SBATCH --ntasks=18表示在一个gpu上,最多运行18个同时并行的任务。这里的18是通过实验得到的,可能会有变动。如果这里设置的任务过多,即使cpu和gpu的资源充足,也无法运行。可能是和服务器的设置有关
#SBATCH --cpus-per-task=4表示每个任务最多分配4个CPU核心,这个值也是通过实验得到的,可能会有变动。如果一个任务分配过多的cpu核心,也会导致无法运行。可能和服务器的设置有关
不过在绝大多数情况下,只要分配合理,使用的资源是不会超出这些上限的,在使用多gpu进行运算的情况下,每一个gpu并不会分配到这么多资源。放心大胆使用即可
# 激活虚拟环境
source /public/home/zht/python39venv/bin/activate
# 切换到存放Python脚本的目录
cd /public/home/zht/mycode
n=20
for ((i=1; i<=n; i++))为了方便,可以在脚本里面设置激活虚拟环境,并且自动切换到存储.py文件的目录,这样就不用每次连接到服务器就手动激活环境以及切换目录了
下面的n以及for就是进行任务的循环
for ((i=1; i<=n; i++))
do
srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN051.py &
srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN052.py &
srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN053.py &
srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN054.py &
srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN055.py &
srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN056.py &
srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN057.py &
srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN058.py &
wait
donesrun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN051.py &
srun:用于提交作业。--cpu-bind=cores:将每个任务的 CPU 绑定到单独的核心上。
--gres=gpu:1:这个选项请求每个任务分配一个 GPU 资源。这里总共有8个srun,即表明总共请求8个gpu。注意:这里如果你把请求的gpu数量设置成了2,那么是请求两个gpu来共同运行你的一个任务,而不是分别运行一个任务。这样如果python代码里面没有进行调整,那么会导致性能的大幅下降。
--ntasks=9:指定了要启动的任务数量。这里总共有8个srun,即总共启动9*8=72个任务。
--cpus-per-task=1:指定了每个任务应该分配的 CPU 核心数。在这里,每个任务将分配一个 CPU 核心,即总共使用72个cpu核心,刚好达到服务器节点的cpu核心数量上限。
python CNN051.py:这是实际要执行的命令,即运行名为 CNN051.py 的 Python 脚本。
&表示在后台运行作业。避免前面的任务阻塞后面的任务。
总结一下,上述这些代码的作用就是使用了一个gpu节点的八个gpu,每个gpu上面分配9个相同的任务,例如第一个gpu上面并行运行CNN051.py九次,例如第二个gpu上面并行运行CNN052.py九次,以此类推。这一整个流程总共执行n次。
可以根据你的机器学习代码的实际网络结构大小和问题规模,结合后面提到的资源监控的部分,来调整这里的并行数量以及cpu资源分配等内容。
python代码设置
只修改脚本配置而不修改机器学习相关的代码是不能实现并行的!
这里以pytorch为例,说明在代码里面如何设置并行。如果你使用的是其他机器学习框架,例如tensorflow等,也需要在代码里面进行类似的设置,可自行查阅资料设置
当你的神经网络模型比较小时(例如只有几层卷积层以及附带的池化层以及少量全连接层)时,不需要利用到每个gpu所有的计算资源,此时建议考虑在单个gpu上面并行多个任务。例如之前示例中演示的,每个gpu上面并行运行九个任务。
那么这种情况下,在代码里面调整非常容易,只要在定义好模型之后,使用torch里面自带的DataParallel,即model = nn.DataParallel(model) ,这样框架就会自动寻找你所配置的资源需求,并且进行并行运行
model = CNN_64(num_classes)
model = nn.DataParallel(model)在python代码设置好之后,执行例如srun --cpu-bind=cores --gres=gpu:1 --ntasks=9 --cpus-per-task=1 python CNN051.py & 的命令,这句命令就表明要寻找一个gpu,然后在上面复制九份进行运行,每一份分配一个cpu核心
同时要在代码里面指定使用gpu运行,否则可能会默认使用cpu运行,使用类似如下的代码指定使用gpu运行:
device = torch.device("cuda" if torch.cuda.is_available() else "cpu")
model.to(device)注意
在你不确定你的python代码是否设置了对应的多gpu配置的时候,尽量不要设置例如一个任务使用多个gpu,例如
srun --gres=gpu:2 --ntasks=1 --cpus-per-task=2 python CNN.py
在这种情况下,并不会创建两个任务放在两个gpu上并行运行,而是会使用两个gpu同时运行一个任务。可能会造成资源的浪费,而且如果模型过小,I/O或者数据在gpu之间调度之类的时间会大量占用时间,造成效率下降。
并行相关的部分
如果你的模型很大?一定需要使用多个gpu并行?并且在多个gpu之间通信传递数据?
也需要调整你的python代码,可以使用类似nn.parallel.DistributedDataParallel之类的方法处理你的代码,让它完成在多gpu下训练的功能
更多的内容请自己进行探索!可以询问gpt,它在大多数情况下都是很好的老师
查看作业运行和资源使用情况
在理想的情况下,在python代码和脚本文件都设置正确之后,就可以进行大规模的并行运算了!
那么我提交并行任务之后,怎么查看我的任务在正常运行,正常利用所有资源呢?或者我怎么确定当前配置下cpu和gpu的分配是合理的?,cpu或者gpu没有成为计算的瓶颈呢?
可以通过squeue的方式简单查看当前作业是否在运行,例如这里显示R表明正在运行
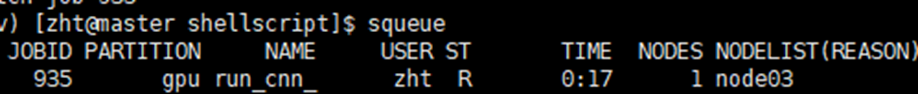
要查看某个节点的资源使用,首先需要连接到该节点。这是后面一切操作的前提
使用ssh+编号进行连接,例如这里的ssh node04 连接成功之后应该会显示当前处于哪个node节点
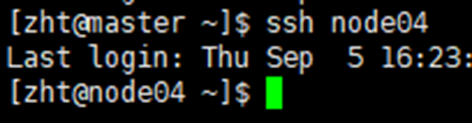 查看gpu资源的使用
查看gpu资源的使用
连接到你需要查看的节点之后,使用命令nvidia-smi进行gpu资源的查看
例如输入命令后,打印的内容如下方所示,显示当前使用了8个gpu,每个gpu的显存使用情况以及gpu的利用率。这里可以看到每个gpu都被合理利用起来了
nvidia-smi命令还会显示运行的进程的若干内容,例如所在的gpu编号,任务路径等(此处未展示)
查看cpu资源的使用
连接到你需要查看的节点之后,使用命令top显示系统中正在运行的进程和系统的资源使用情况,然后按1查看cpu的使用情况。
例如这里显示所有的cpu使用率都接近100%,说明资源正常使用。一个节点总共有72个cpu核心,但是这里只显示了34*2=68个,原因未知。但是不影响正常使用,执行分配还是可以分配72个cpu核心
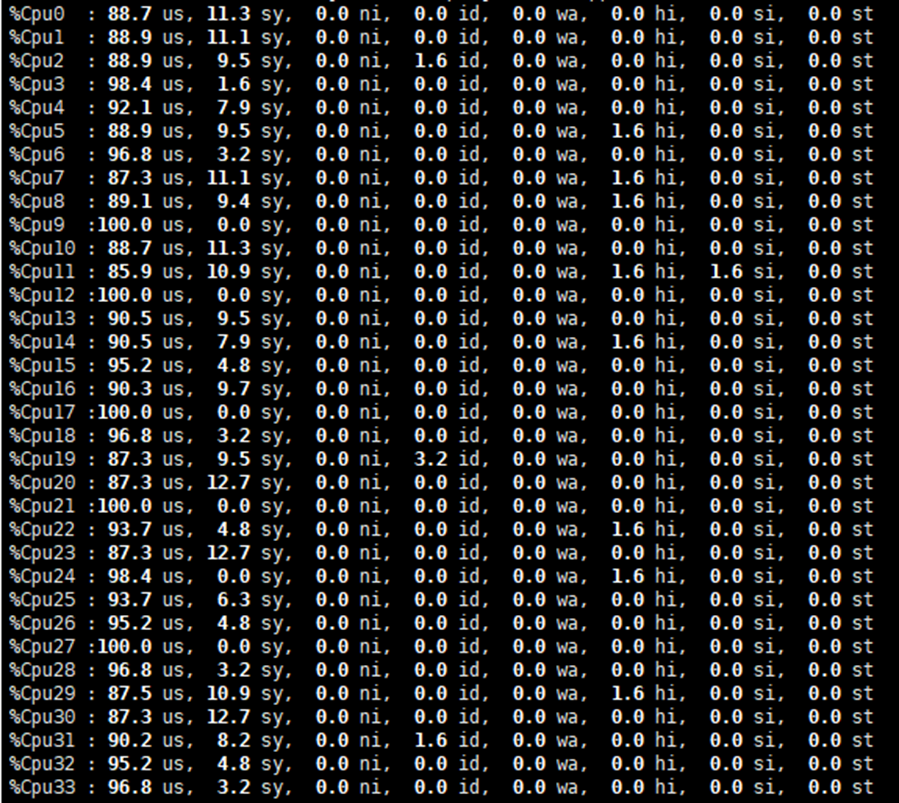
合理分配cpu和gpu资源
以当前使用的神经网络举例,说明资源分配相关问题。不同的神经网络面临的情况不一样,需要进行灵活变通:
由之前的两个图我们可以得到,在当前配置下,cpu的利用率大约是90%+,而gpu的利用率大约是60%,这说明gpu资源更有富足。
在当前的神经网络设置下,根据测试,在小规模神经网络高并行的情况下,限制性能更多的还是在于cpu。测试中把每个任务分配的cpu核心从1提升为2,性能会获得不错的提升。
例如使用4个gpu,每个gpu并行执行9个任务,每个任务分配2个核心,比使用4个gpu,每个gpu并行执行9个任务,每个任务分配1个核心要快很多。
但是在当前神经网络的设置的情况下,和gpu资源相比,当前cpu核心还是偏少,因此同样使用全部的72个cpu核心,在当前神经网络情况下,更推荐使用全部的8个gpu,每个gpu并行执行9个任务,每个任务分配1个核心。这样单个任务的平均时间会下降。
后续配置自己的神经网络的运行脚本时候,可以把先服务器当作gpu资源更加充足的情况,进行脚本的设置
结语
本文档中大部分结论基于实验测试,可能有表述不准确乃至错误的地方,如果在阅读文档的时候出现疑问,可以上网搜索相关内容或者查看一些学校自己的slurm服务器使用教程进行进一步的理解,例如上交的超算平台手册之类的内容https://docs.hpc.sjtu.edu.cn/job/slurm.html
希望这篇文章对你有帮助!

